


2026-01-05
有临医药

引言
放射性标记人体物质平衡研究是创新药临床药理学研究的基石,被视为获取药物在人体内吸收、分布、代谢和排泄(ADME)全面定量信息的“最直接”方法。该研究通过追踪放射性示踪剂,能够无偏倚地确定药物的整体代谢与排泄途径、鉴定循环代谢物并评估其相对暴露量,从而为后续临床开发提供关键依据。近年来,全球主要监管机构相继更新了相关技术指导原则,标志着此类研究的规范进入新阶段。例如,中国国家药品监督管理局药品审评中心(CDE)于2024年发布了《放射性标记人体物质平衡研究技术指导原则》,美国食品药品监督管理局(FDA)也在2024年发布了《Clinical Pharmacology Considerations for Human Radiolabeled Mass Balance Studies Guidance for Industry》最终行业指南。这些法规与指南共同构成了当前研究的核心框架。本文旨在结合国内外最新监管要求,系统综述放射性标记人体物质平衡研究的关键设计要素,以期为研究的规范化设计与实施提供参考。
一、放射性标记人体物质平衡研究的核心价值
放射性标记人体物质平衡研究是新药临床药理评价体系中不可或缺的关键环节。它通过提供一份关于药物在人体内吸收、分布、代谢和排泄(ADME)的 “全景图” 与 “定量账本” ,为科学认知、监管决策与研发策略提供了任何非放射性技术无法完全替代的坚实数据基础。其核心价值主要体现在以下三个方面。
1.提供全景式与定量化的ADME数据,确立认知黄金标准
该研究凭借放射性示踪技术,能够无偏倚、全谱系地追踪所有源自母体药物的物质(包括未知代谢物),实现对药物体内吸收、分布、代谢和排泄(ADME)全过程的无遗漏解析与直接定量。与传统依赖已知对照品进行靶向分析的液相色谱-质谱联用(LC-MS)技术相比,放射性示踪法能系统而可靠地回答“药物在体内去了哪里”这一根本问题,精确测定药物及其相关物质在血液、尿液及粪便中的回收率与动力学特征,从而成为全面理解药物处置过程的黄金标准。流程如Fig 1 所示。
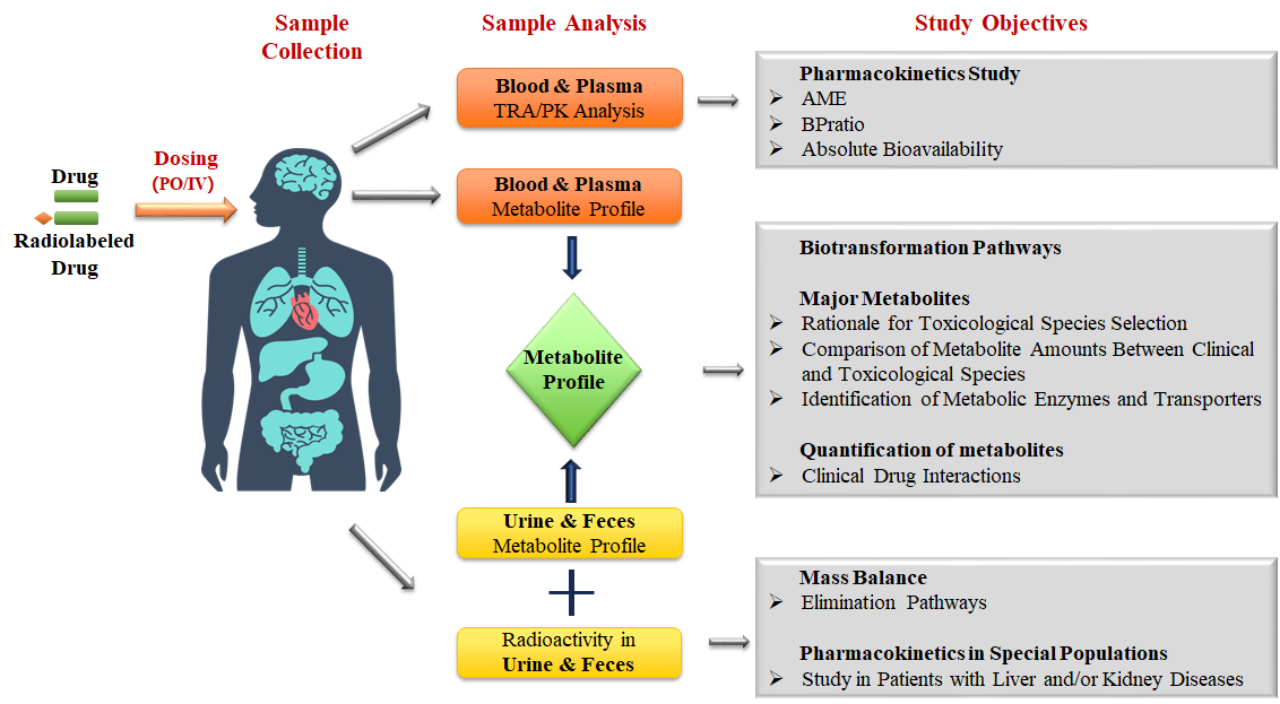
Fig 1. Radiolabeled Human Mass Balance Studies Work Flow
2.支撑关键监管决策,是新药注册的基石
研究数据直接关联并强力支撑两项核心监管评估,是监管机构(如FDA、NMPA、EMA)要求新药申请必须提交的关键组成部分,其价值主要聚焦于以下两大评估维度:
准确定量人体循环中代谢产物的暴露水平。若其暴露量超过总药物相关物质暴露量的10%(特定阈值),则需启动针对性的非临床安全性研究,以评估潜在风险。决策树如Fig 2 所示。
明确药物的主要排泄途径(如肾排泄或肝胆排泄)。该信息直接决定了是否需开展肝、肾功能不全患者的专项药代动力学研究,并对临床药物相互作用的风险评估具有至关重要的指导意义。
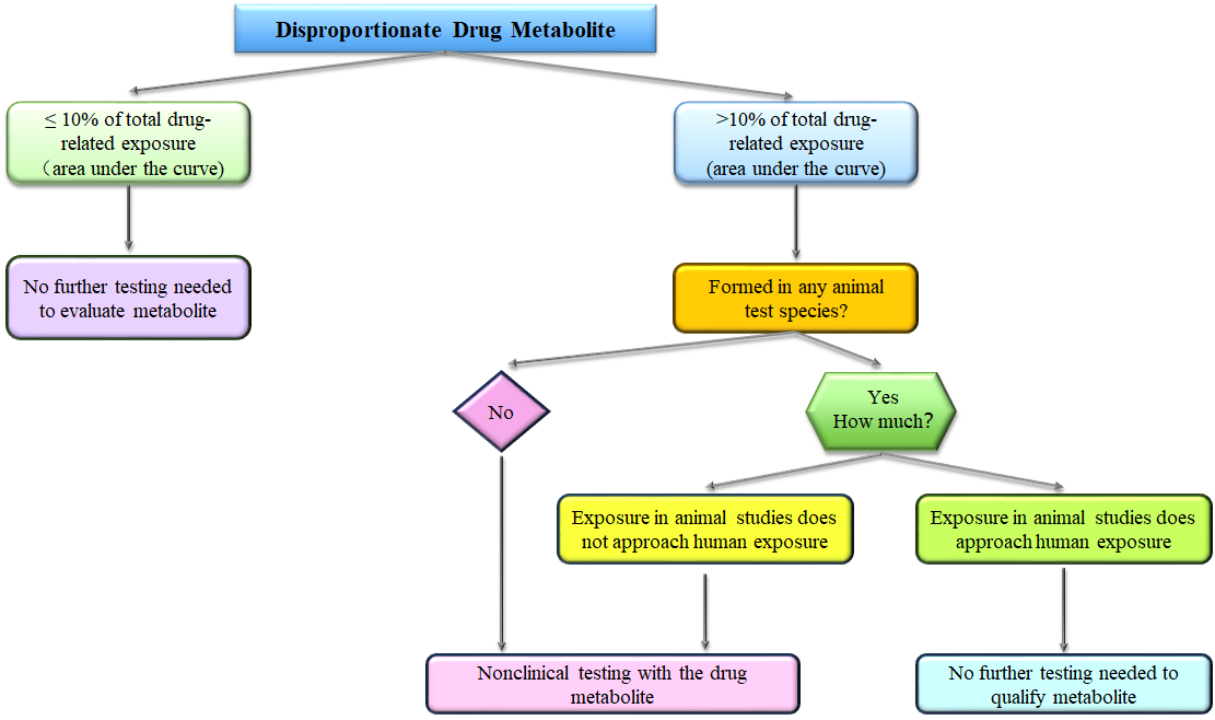
Fig 2. Decision Tree Flow Diagram
3.赋能研发决策,提升成功率并管控风险
在临床开发早期(通常于III期确证性试验前)获得完整的人体物质平衡数据,能够帮助研发者及早洞察潜在的代谢缺陷、不理想的清除特性或高比例活性/毒性代谢物等问题。这使得企业能够基于可靠的人体数据,对研发项目做出更明智的 “继续/终止” 决策,避免将存在根本性药代缺陷的化合物推进至耗资巨大的后期临床试验。该研究通过前瞻性地识别关键风险,显著优化了资源配置,降低后期研发失败的概率,最终提高整体研发效率与成功率。
二、适用范围
一般情况下应对所有的新分子实体药物开展放射性标记人体物质平衡研究,除满足以下任意一种情况:
三、开展时机
《放射性标记人体物质平衡研究技术指导原则(征求意见稿)》中明确指出,鼓励在药物临床开发的早期来进行放射性标记人体物质平衡研究,建议在III期前或者确证性临床研究开始前完成,最晚也须在NDA之前完成。放射性标记人体物质平衡研究在III期前完成可以支持III期临床研究的设计,例如肝/肾损伤患者是否纳入研究、合并用药的纳入与避免等。放射性标记 ADME 研究的通用实施框架如 Fig 3 所示。
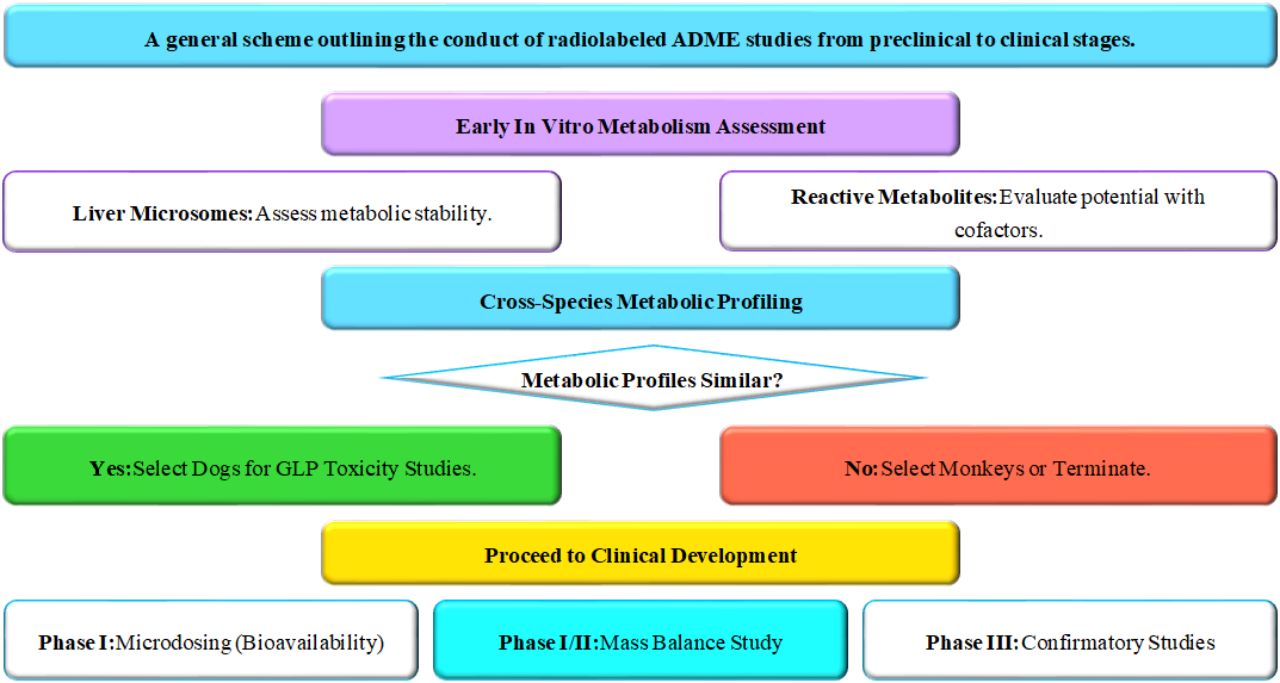
Fig 3. General Scheme for Radiolabeled ADME Studies
四、研究设计的关键考量要素
放射性标记人体物质平衡研究的顺利实施与成功推进,高度依赖于科学严谨且周密完备的研究设计。下文将结合当前科研实践进展与监管技术要求,对该研究的各项关键要素展开详细阐述。
1.放射性同位素的选择
在放射性标记ADME研究中,碳-14(14 C)是绝对主流且受到推荐的选择,占据了全球范围内99%以上的研究。其优势在于:半衰期极长(约5730年),在研究期间无需进行放射性衰变校正;释放的β射线能量低、穿透力弱,生物安全风险相对可控。14 C原子能够稳定地嵌入有机分子的骨架结构中(如羧基、甲基),在体内代谢过程中不易丢失,从而确保了对药物主要代谢路径的完整追踪。
相比之下,氚(3 H)虽然成本较低且允许更高的比活度,但其应用存在显著局限性:3 H原子易与体液中的氢发生交换,导致标记丢失;C-3 H键可能因同位素效应而改变代谢速率;其检测灵敏度通常也低于14 C。因此,3 H标记通常仅用于特殊情况,如追踪分子中特定的、稳定的氢原子位置。
2.放射性标记位点的选择
标记位点的选择是放射性标记人体物质平衡研究设计的核心化学考量。总原则是将放射性核素标记在分子结构中代谢稳定性高、不易在体内生物转化过程中脱落的化学位点,通常是分子的芳香环或饱和碳骨架等“代谢惰性”区域。这确保了放射性信号能够代表母体药物及其主要代谢产物的去向。
3.制剂制备与质量控制
为确保给药剂量的准确性和代表性,放射性标记制剂必须满足严格的质量标准:
4.给药方案设计
给药途径:原则上需与药物临床拟用途径一致。对于吸入剂等操作复杂或难以定量回收的给药方式,可在充分科学论证后采用替代途径(如静脉注射)进行研究,但需在报告中明确阐述替代方案的局限性及对研究结果解读的影响。
给药频次:单次给药是标准设计,适用于绝大多数药物。当药物存在明显的时间依赖性药代动力学特征(如自身酶诱导/抑制效应),或其安全性窗口较窄,需要长期连续用药才能在患者体内达到稳态时,则需考虑采用多次给药至稳态后的物质平衡研究设计,以更真实地反映临床用药场景下的药物处置特征。
剂量确定:
5.受试者选择与伦理考量
人群选择:尽管约90%的研究选择健康成年男性志愿者以最大程度减少变异性,但在以下情况下,应优先或必须选择目标患者人群:
样本量:依据中国国家药监局药品审评中心(CDE)、美国食品药品监督管理局(FDA)等监管机构的技术指南,放射性人体物质平衡研究的最低有效样本量通常为 6 例;若研究为探索个体间高变异性的微剂量研究,或针对特定患者群体开展,则建议将样本量扩大至 8~12 例,以提升数据的代表性与统计稳健性。
6.生物样本采集、处理与分析策略
样本采集无需遵循固定时间方案,而是基于预设的放射性终止标准,以实现个体化、高效的试验流程。
血液/血浆样本:
排泄物样本:
7.放射性分析技术平台
放射性分析技术的选择与应用流程如 Fig 4所示。通过对上述要素进行科学与合规的精细化设计,放射性标记人体物质平衡研究才能可靠地生成关于药物体内命运的黄金标准数据,为新药开发的关键决策提供坚实支撑。
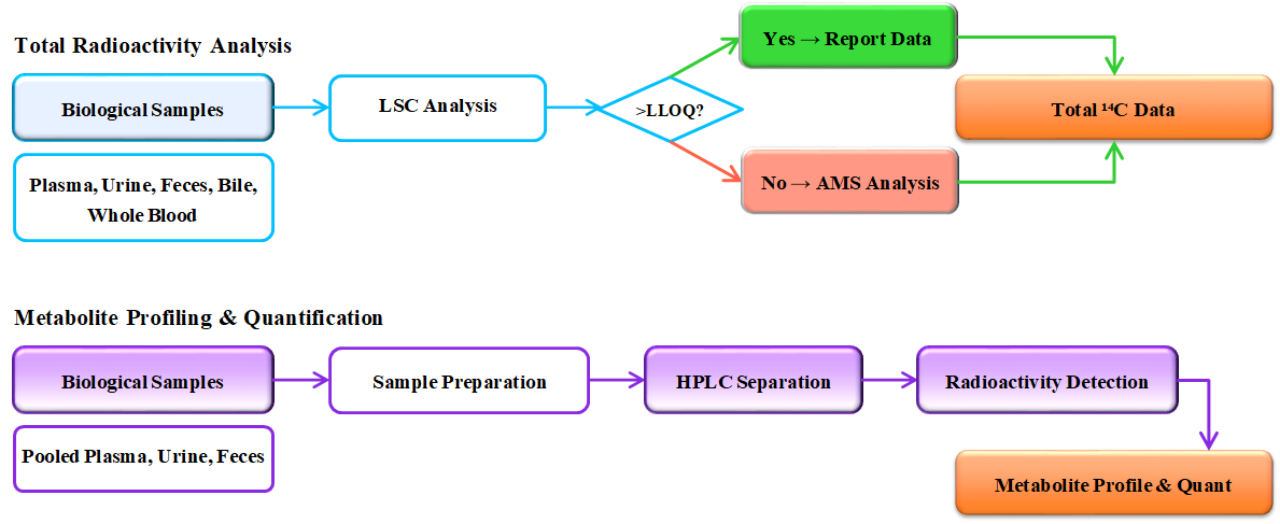
Fig 4. Analysis Workflow for Radiolabeled ADME Studies
五、结语
国内放射性同位素示踪技术在药物临床研究中的应用已走过十余年发展之路。CDE《放射性标记人体物质平衡研究技术指导原则(征求意见稿)》的出台,进一步明确了同位素示踪研究在药物人体评价中的核心地位,使其成为驱动新药临床研发创新发展的关键趋势之一。对于相关科研与技术人员而言,需兼顾新药临床研发整体策略的规划与落地,更应着力推动临床研究技术方法与检测平台的突破创新;尤为重要的是,优质、高效的放射性标记人体物质平衡研究设计,能够精准助力核心研发决策、规避研发风险,从而显著加速药物研发进程。未来需以科学规范的临床药理学设计为依托,充分释放同位素示踪技术的应用价值,提升新药研发的科学性与高效性。
参考文献
[1] 国家药品监督管理局药品审评中心。放射性标记人体物质平衡研究技术指导原则. 2024.
[2] FDA. Clinical Pharmacology Considerations for Human Radiolabeled Mass Balance Studies Guidance for Industry. 2022.
[3] ICH. ICH S3A: Note for Guidance on Toxicokinetics: The Assessment of Systemic Exposure in Toxicity Studies. 1994.
[4] FDA. Safety Testing of Drug Metabolites. 2020.
[5] Radiolabeled Absorption, Distribution, Metabolism, and Excretion Studies in Drug Development: Why, When, and How? Natalia Penner, Lin Xu, and Chandra Prakash. Chemical Research in Toxicology.2012
撰写:佟巍
审核:谢珊珊、韩海雄










