


2026-06-15
引言
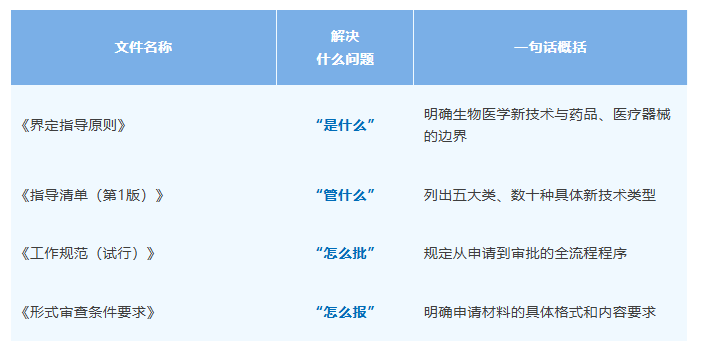
2026 年 4 月 30 日,国家卫生健康委发布了《生物医学新技术与药品、医疗器械界定指导原则(暂行)》、《生物医学新技术临床转化应用审批工作规范》两份法规以及配套的《生物医学新技术临床研究备案指导清单(第 1 版)》、《生物医学新技术临床转化应用申请形式审查条件要求》等文件,共同构建我国生物医学新技术从临床研究到临床转化应用的全链条监管框架。这标志我国在生物医学新技术领域迈出里程碑式的一步。
今天,我们就上述四份文件的核心内容做一下解读。
01
四份文件,各司其职
02
《界定指导原则》:先搞清楚“这是什么”
这份文件是四份文件中的“总纲”,核心任务是划清生物医学新技术与药品、医疗器械的边界。
四大总体思路
1.统筹发展和安全——聚焦于难以开发为药品或医疗器械的新技术,在保证安全底线的前提下支持发展。
2.满足人民群众健康需求——聚焦于为尚无有效治疗手段的疾病提供新的治疗方向。
3.充分考虑不同阶段特点——对临床前研究、临床研究和转化应用等不同阶段,采用不同界定方式。
4.注重部门间政策衔接——实现与药械管理体系的“错位发展”与功能互补。
两个阶段的界定方式
03
《指导清单》:管什么?五大类新技术一览
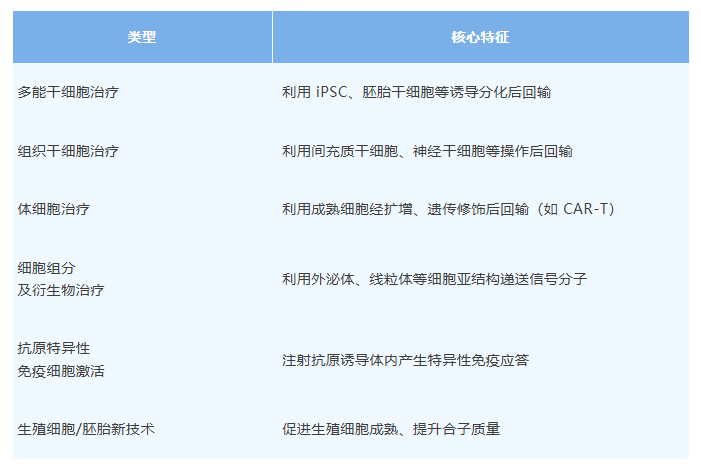
《指导清单(第 1 版)》将生物医学新技术分为五大类,涵盖当前最具创新性和监管挑战性的前沿领域。
1.基因治疗新技术(4类)

2.细胞及其衍生物治疗新技术(6类)
3.组织器官治疗新技术(2类)
4.微生物治疗新技术(3类)
5.脑机接口新技术(2类)
04
《工作规范》:怎么批?核心审批流程详解
《工作规范》共五章二十六条,是审批工作的核心操作指南。以下是最值得关注的几个重点:
1.哪些技术需要审批?
已列入《指导清单》的技术,满足以下条件之一方可申请审批:
高度个性化:国内尚无同类机制原理的药品上市或启动确证性临床试验
治疗罕见病:国内尚无同类机制原理、针对相同适应症的药品上市或启动确证性临床试验
注意:符合医疗器械定义的,按医疗器械法规注册,不纳入本审批范围。
2.申请需要满足什么条件?
必须同时满足三个条件:
已完成经备案的临床研究
临床研究证明技术安全、有效、符合伦理
经多中心参与,且能获得一致的安全性、有效性结论
3.审批流程是怎样的?
申请提交 → 形式审查(5 个工作日) → 材料核查 → 技术评估 + 伦理评估 → 专家提出评估意见 → 国家卫健委 15 个工作日内作出决定
评估结论有四种:
建议批准——技术安全有效、成熟稳定且符合伦理
建议不予批准——技术不成熟、证据不足、风险大于获益、存在伦理风险等七种情形
延期再审——需补充材料
终止审查——存在重大问题或不属于审批范围
4.有快速通道吗?
有!用于治疗严重危及生命且尚无有效治疗手段的疾病,或公共卫生方面急需的新技术,可以申请优先审查审批,启动优先评估程序。
5.批准后怎么管理?批准后,不同风险等级的技术有不同期限的“独占期”:

再评估机制:当出现安全性、有效性认知改变、严重不良反应或重大社会风险时,国家卫健委将组织再评估,期间暂停应用。再评估不合格的,禁止临床应用。
05
《形式审查条件要求》:怎么报?材料准备要点
这是申请材料能否被受理的“第一道门槛”,核心要求如下:
申请机构资格条件
境内依法登记、具有独立法人资格的实体
必须作为发起机构完成备案临床研究
无科研严重失信、严重医疗质量不良事件记录所有参与确证性临床研究的机构均需完成GCP备案
技术负责人资格条件
必须是申请机构的正式聘用人员
应具有高级职称或同等技术能力
无失信记录并签署诚信承诺书
申报材料清单(13 项必须提交)

视情况提交的材料:既往审批情况、知识产权说明、回避事项申明、人类遗传资源备案材料、优先审查审批申请表等。
技术命名规范
建议按照 “核心干预手段+目标细胞/靶点+预期用途” 格式命名。
示例:“慢病毒转导的 NY-ESO-1 靶向自体 TCR-T 细胞治疗晚期滑膜肉瘤技术”
06
总结:新规带来的四大变化
未来展望
该监管体系的实施,将有效引导生物医学新技术从“实验室”向“病床”的合规转化。对于相关机构而言,深入理解文件的内在逻辑和具体要求,是成功将前沿技术转化为临床价值的关键前提。随着技术发展和监管实践经验的积累,该体系将进行动态调整,以适应生物医学领域的快速创新。











